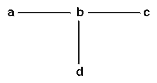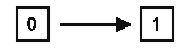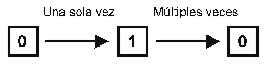|
|
Metodos de analisis en la reconstruccion filogenetica. Bol. SEA, 26, 1999 : 45-56. |
|
|
Métodos de análisis en la reconstrucción filogenética Emilio J. López Caballero 1 y Gonzalo Pérez Suárez 1
1.- Breve historia de la Clasificación. Los caracteres taxonómicos Mientras que todas las ciencias se encuentran hoy día en una situación comprometida debido a la multiplicidad y cantidad de información, las ciencias biológicas han estado en esa situación desde hace mucho tiempo. Esto se debe a que muchos organismos se encuentran en muy variadas situaciones y porque gran cantidad de ellos han atraído la atención del hombre. Bastante antes de la invención de la escritura y, por ello, del comienzo de la historia, el hombre prehistórico debió haber acumulado un conocimiento enciclopédico de su medio orgánico en términos de qué era vivo y porqué, qué era amigable o enemigo, alimento o veneno. La existencia de conceptos tales como alimento, implica que los organismos se han agrupado y esto implica una clasificación. Simpson (1961) seńaló que la necesidad de juntar seres en categorías es una característica general de los organismos vivos e indica que Amoeba, de forma semejante al hombre, no puede existir sin la apreciación de la categoría “alimento”. El pensamiento humano, en general, como se refleja en el lenguaje, parece depender fuertemente del reconocimiento de grupos, particularmente los grupos de objetos a los que damos nombres colectivos. Tenemos que notar que el nombre “insecto” implica dos procesos mentales, primero el de considerar, de forma “subconsciente”, las semejanzas que poseen “todos” los insectos y segundo, considerar las diferencias entre los “insectos” y los “no insectos”. La necesidad de una clasificación taxonómica fue seńalada convincentemente en 1970 por Savory: “encontramos animales tan diferentes en el mundo que no podemos tratarlos separadamente e, incluso, si quisiéramos hacerlo, la tarea estaría más allá de la capacidad de la mente y de la memoria humana. La clasificación nos viene impuesta por las limitaciones del cerebro”. En el contexto en que se expresa Savory y en el de muchos otros autores (Mayr et al., 1953, p. e.), el concepto de clasificación biológica está restringido al agrupamiento de organismos en taxones - desde el Phylum hasta el género y la especie - por sus atributos estructurales.
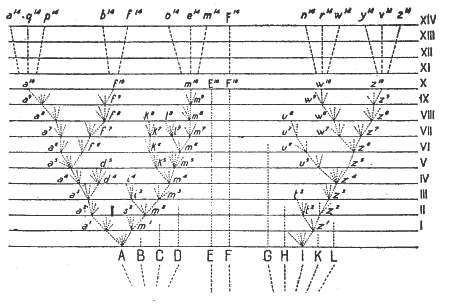
Los taxónomos empíricos no pueden proporcionar una explicación causal para el hecho de ser capaces de agrupar las especies de acuerdo con “relaciones” o “afinidades”. Cuando Strickland (1840) definió afinidad como “la relación que hay entre dos o más miembros de un grupo natural, o en otras palabras, una coincidencia en caracteres esenciales”, dejó las palabras clave, “natural” y “esencial”, sin definir. Darwin vino a rellenar ese hueco y mostró por qué hay grupos naturales y por qué comparten caracteres “esenciales”. Como seńaló Simpson (1961), hay dos tipos de relaciones que se pueden utilizar para hacer un agrupamiento general, a saber, la “asociación por semejanza” y la “asociación por contigüidad”, siendo conocida la primera generalmente como “sistemática” y la segunda como “taxonomía”. Las definiciones aceptadas de estos términos incluirán, lógicamente, esos conceptos y así la definición de Sistemática sería “El estudio científico de los tipos y diversidad de los organismos y de cualquiera y de todas las relaciones entre ellos”, mientras que la Taxonomía sería “El estudio teórico de la clasificación, incluyendo sus bases, principios, procedimientos y reglas”. Darwin fue por ello, también, quien proporcionó la teoría básica de la clasificación biológica, aunque Linneo sentara unas primitivas bases sobre métodos. Nadie antes de Darwin había seńalado tan inequívocamente que los miembros de un taxón son semejantes porque son descendientes de un ancestral común. Esta idea, no obstante, no era enteramente nueva, ya que Buffon, el propio Erasmus Darwin y algunos evolucionistas alemanes, habían jugado con la posibilidad que especies similares, tales como caballo y asno, o todos los gatos, podían haber derivado de una especie ancestral. Linneo, en sus últimos ańos, había sugerido que los miembros de un taxón más alto pudieran ser producto de hibridación. Ni Linneo ni Buffon convirtieron estas especulaciones en una teoría de clasificación ni de evolución. Muy pocos comprenden que Darwin fue el fundador de todo el campo de la taxonomía evolutiva. Como seńaló correctamente Simpson “la taxonomía evolutiva proviene explícitamente y casi exclusivamente de Darwin”. Pero esto significa que no sólo se explica automáticamente la teoría de la descendencia común para muchos de los grados de semejanza entre los organismos, sino que Darwin desarrolló también una teoría bien fundamentada con una declaración detallada de métodos y dificultades (consultar el capítulo decimotercero de la primera edición del Origin...). Él hace referencia a su famoso diagrama (Fig. 1), en el que cada una de tres especies cogenéricas del Silúrico (A, F e I), tienen descendientes modernos de rango muy diferente. La línea derivada de la especie F ha cambiado tan poco que todavía contiene un único género, mientras que sus dos grupos hermanos, A e I, constituyen, en la actualidad, diferentes familias o incluso ordenes. Para la determinación del “rango” Darwin, generalmente, aplicó el principio del grado de divergencia, más que el de proximidad, al punto de ramificación. En particular, rechazó la idea, tan ampliamente extendida entre los botánicos de los siglos XVII y XVIII, y también entre los zoólogos, desde Cuvier, de que cuanto más importante es una estructura para la supervivencia y la perpetuación de un organismo, más importante es también esa estructura para su clasificación, lo que ilustra con varios ejemplos como el del valor altamente desigual que tienen las antenas como un carácter taxonómico en diferentes familias de insectos y para rechazar aquella idea seńala explícitamente: “Que la mera importancia fisiológica de un órgano no determina su valor para la clasificación se puede demostrar por el hecho de que en grupos relacionados, en los que el mismo órgano... tiene prácticamente el mismo valor fisiológico, su valor para la clasificación es ampliamente distinto”. El consejo de Darwin no constituye un rechazo de la importancia de la selección natural, lo que dice es que las adaptaciones especiales pueden implicar solamente una porción limitada de la dotación genética de un grupo y por ello ser menos informativas que, p. e. el “hábito” general. Además, las adaptaciones especiales pueden ser adquiridas independientemente en varias líneas evolutivas no relacionadas, o sea que están sujetas a convergencia. Para la evaluación de los caracteres el propio Darwin propone ciertas reglas. Considera de elevada importancia taxonómica aquellos que son constantes en varios grupos grandes. Llama la atención sobre los errores que pueden cometer los taxónomos al considerar “semejanzas espúreas” causadas por evolución convergente (semejanzas análogas o adaptativas). Otras dos recomendaciones son particularmente importantes, como es el rechazar las semejanzas debidas a una descendencia de semejanzas superficiales producidas por convergencia, y la otra se refiere al “peso” de los caracteres. Esta segunda es importante ya que algunos caracteres tienen un contenido de información mayor que otros. Este peso viene definido por la correlación de ese carácter con las partes de la clasificación establecidas de manera más firme (por varios métodos de comprobación). En la actualidad se combina este método con varias técnicas de computación para determinar esa importancia (“peso”). En lo que se refiere a la metodología de la clasificación, la revolución darvinista sólo tuvo un pequeńo impacto. Es evidente que el punto crucial para el giro en la historia de la taxonomía fue el abandono del esencialismo y de la “clasificación descendente”, ocurrido antes de 1859. La decisiva contribución hecha por Darwin a la taxonomía fue doble: por su teoría de la descendencia común que proporcionó una explicación de la existencia de la jerarquía linneana, y por la homogeneidad de los taxones en una clasificación “natural”, restaurando el principio de continuidad entre los grupos de organismos rechazado por Cuvier y por los Naturphilosophen alemanes en su teoría de los arquetipos. A partir de 1880 hubo un declive, gradual y visible, en cuanto al interés por los estudios taxonómicos y filogenéticos, debido a varias razones. Quizá el factor más importante para este declive fuese la competencia creciente de otras ramas de la biología experimental en las que se realizaron excitantes descubrimientos como la citología, genética mendeliana, fisiología, bioquímica, etc., que condujeron a los biólogos más jóvenes hacia esos campos de investigación, por lo que el espléndido inicio que hizo Darwin al desarrollar una teoría y una metodología de la taxonomía fue ampliamente ignorado en el período post-darvinista, entrándose en una etapa en la que no se respetaron esquemas anteriores, ni reglas de nomenclatura, ni criterios sobre la validez de los caracteres, etc. (Mayr, 1982, p.220). Hacia la década de 1930 el interés resurgió y fue finalmente estimulado al inicio de los 40 por dos libros de J. S. Huxley, “La nueva sistemática” de 1940 y “La evolución, la síntesis moderna” de 1942. La tesis defendida es que las unidades sistemáticas eran las poblaciones y no los individuos y que las especies no deberían estar ligadas al ejemplar (o ejemplares) tipo definidos por el autor original. Se estudió más intensamente la variabilidad intraespecífica y se propusieron métodos estadísticos para la distinción entre especies y “dentro” de las especies. Ciertamente, gran cantidad de estas ideas no eran realmente nuevas y, por ejemplo, el deseo de conservar grandes series de ejemplares de cada especie, había sido ya aplicado desde hacía tiempo en los museos y sirva de ejemplo de lo seńalado lo que Mayr et al. 1953 recogen de Baird, 1854, relativo a la Smithsonian Institution: “El objetivo de la Institución al hacer sus colecciones no es simplemente poseer las diferentes especies, sino también determinar su distribución geográfica, por lo que es de importancia poseer de cada localidad series tan completas como sea posible...” La obra “La nueva sistemática” fue valiosa con relación a que dirigió la atención hacia los mecanismos de especiación y a que ligó las actividades de los genéticos y biogeógrafos con la corriente principal de la biología sistemática. No obstante la taxonomía falló en relación con recuperar su vigor primitivo y, el impacto creado por “La nueva sistemática” fue menor de lo que sus proponentes habían esperado. Posiblemente el “biólogo medio” se asustó por la estadística, aunque no hubiera necesidad de conocer sus implicaciones y podía ser usada con precaución y de manera simple. Los conocimientos a este respecto se limitaban en ese momento a la interpretación de una curva bimodal, histogramas, diagramas de dispersión, desviaciones estándar, chi-cuadrado y coeficientes de diferencias medias. Parte de la relativa falta de interés por “La nueva sistemática” podía ser debida a un cambio en la racionalidad de los criterios para la identificación de las especies. Pocos parecían interesados en el análisis exhaustivo de las afinidades de los taxones y muchos consideraban la identificación como un medio para otro fin, por ejemplo, en ecología y no hay duda de que este tipo de estudios dependían completamente de los expertos en taxonomía. Chace (1969) seńalaba correctamente: “...un problema importante y, virtualmente insuperable, que arrastra cualquier estudio ecológico de las aguas tropicales hoy día, es el imperfecto conocimiento de la fauna existente...”. En nuestros días, con el fervor por la conservación del medio, de las especies, de la biodiversidad, es fantástico que solamente aquellos investigadores que nos puedan decir qué especie ocupa un determinado medio, sean un pequeńo grupo (y cada vez menor) de taxónomos mal considerados, aunque, eso sí, dedicados con fervor a su trabajo. Quizá el excelente desarrollo constructivo de aquel período fue dar un significado ecológico a los taxones más elevados. Se reconoció que los taxones más altos estaban formados por especies que, en su conjunto, ocupaban un nicho específico o una “zona adaptativa” (en palabras de Mayr, 1982). Como “La nueva sistemática” se concentró ampliamente sobre el nivel específico, no ofreció soluciones a las necesidades de la macrotaxonomía y la ayuda tenía que venir de otra parte. De manera independiente se propusieron dos soluciones drásticamente distintas: la Fenética numérica y la Cladística. Ninguna de ellas se propuso como reforma a lo que había sino como un reemplazo revolucionario de los procedimientos existentes, manteniendo su vigencia la Metodología evolutiva o tradicional, desarrollada sin solución de continuidad por una escuela y cuyos principios se pueden encontrar en Mayr (1969). Antes de realizar una breve descripción de cada una de estas aproximaciones al conocimiento de la estructura de las relaciones en el mundo de los seres vivos, veamos los caracteres taxonómicos, tradicionales y nuevos, y qué uso hacen de ellos estos tres métodos de clasificación. La base de estos tres métodos es el análisis y la evaluación de los caracteres taxonómicos. Una insuficiencia de caracteres informativos es la razón más frecuente de porqué no se puede resolver la falta de coincidencia entre las clasificaciones propuestas por uno u otro método. No deja de sorprender que la queja más frecuente que expresa un taxónomo, es que el grupo de animales con los que trabaja no presenta suficientes caracteres para permitir una decisión inequívoca sobre sus relaciones. Dos fenómenos contribuyen a esta dificultad, primero el hecho, bien conocido, de que el fenotipo, en determinados grupos de organismos, está notablemente “estandarizado” (como en los cientos de especies de Rana o en los miles de especies de Drosophila) y por ello proporciona solamente unos pocos indicios morfológicos de las relaciones, y segundo, que una desviación del tipo estándar generalmente sólo afecta a un complejo funcional simple, correlacionado con alguna adaptación especial “ad hoc”. El cambio a una nueva fuente de alimento o la adopción de un nuevo conjunto de seńales de cortejo, puede, a veces, tener como efecto una notable adquisición morfológica que puede ser dividida en un número considerable de caracteres individuales. Considerar éstos como caracteres separados nos podría conducir a error, puesto que hablando en términos filogenéticos, son puramente un reflejo de un único cambio funcional. Darwin ya alertó frente a muchas de las especializaciones “ad hoc”. Una dificultad hallada por los taxónomos con mucha frecuencia está representada por el conflicto que se establece entre las conclusiones basadas sobre diferentes estructuras. El estudio de las extremidades puede indicar que un taxon b está más claramente relacionado a otro a, mientras que los rasgos del tracto intestinal sugieren que el taxón b está más próximo al taxón c. En estos casos el estudio de rasgos adicionales de las extremidades o del tracto intestinal raramente proporcionan una solución satisfactoria. Se pueden encontrar numerosos ejemplos de este tipo de casos en cada uno de los taxones más elevados y debido a ello, los taxónomos, en las últimas décadas han dedicado gran atención a la búsqueda de nuevos caracteres taxonómicos. Aunque los análisis morfológicos cuidadosos proporcionan continuamente nuevos caracteres, los caracteres no-morfológicos juegan un papel, que se incrementa día a día, en el establecimiento de clasificaciones y entre ellos cabría seńalar caracteres tales como componentes del comportamiento, ciclo de vida y anual, fisiología, ecología (por ejemplo, utilización del nicho), parásitos y cualesquiera otros atributos de cada organismo. Muchos de ellos son útiles para la discriminación de especies y otros son indicativos de relaciones entre grupos de especies. La distribución geográfica proporciona, a menudo, pistas inesperadas. La búsqueda del grupo relacionado más próximo en un área geográfica fácilmente accesible no se puede utilizar en el caso de algunos grupos con unas facilidades de dispersión muy elevadas o en grupos que han quedado relegados a áreas muy restringidas, pero sí es muy útil en otros casos (como ha demostrado Simpson en numerosas ocasiones en los animales). Una combinación de análisis cladísticos y biogeográficos, como demostró Hennig y sus seguidores, a veces es particularmente reveladora. El desarrollo tecnológico está permitiendo, en la actualidad, llegar a límites insospechados en la estimación de la estructura fenética de un organismo. Como ejemplo podemos citar la microscopía electrónica de barrido (Scaning) que permite apreciar microestructuras en relieve con una gran capacidad de definición, y la gran variedad de técnicas bioquímicas para medir indirectamente la “distancia genética” entre los organismos. Estas técnicas han alcanzado gran popularidad entre los sistemáticos y en ellas se incluyen la hibrización de ADN, eletroforesis, electroenfoque, isoelectroenfoque, cromatografía líquida de alta presión (HPLC), análisis de las actividades enzimáticas e índices de semejanza inmunológica, etc. que permiten detectar los polimorfismos de proteínas existentes en un órgano o estructura de un organismo determinado. Estos y otros tipos de datos (recogidos de forma más general en Tabla I) son utilizados a diario por los taxónomos. La base fundamental para la anatomía comparada (o la bioquímica comparada, o la etología comparada, etc.) es el concepto de homología. Veamos en un cuadro comparativo algunas de las características generales de los tres métodos de clasificación y el uso que cada uno hace de una serie de atributos. Una relación, no exhaustiva, de los diferentes tipos de caracteres taxonómicos se recoge en la Tabla I.
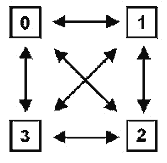
En la Tabla II se pueden comprobar las relaciones entre los tres principales sistemas de clasificación, atendiendo a la consideración que cada uno de ellos tiene para algunos de los tipos de caracteres/atributos que podemos considerar en los taxones. 2.- Breve descripción y comparación crítica de los tres sistemas de clasificación La Fenética, denominada a veces también como Taxonomía numérica o Taximetría, se desarrolló como consecuencia de la disponibilidad creciente de computadores al final de los ańos 50. La base filosófica para la “fenética numérica” es el argumento de que como nunca es posible conocer con certeza cual de las diferentes filogenias en competencia es la correcta, la descripción real de los individuos de un grupo ayudará a conocer la evolución de ese grupo, pero nada más. Por lo anterior, los organismos deberían ser clasificados estrictamente en función de la conveniencia, en lugar de establecer clasificaciones basadas sobre reconstrucciones hipotéticas de la historia filogenética de un grupo de animales o plantas, o sea, como los libros en una biblioteca. Los “fenetistas” analizan los datos mediante uno o más programas de ordenador que harán que se agrupen las OTU’s (Unidades Taxonómicas Operativas, concepto introducido por Peter H. A. Sneath y Robert R. Sokal, 1973) en función de índices de semejanza, o de diferencia, global y producen un diagrama ramificado (dendrograma, fig 2) denominado fenograma. Este se puede transformar en una clasificación y esta transformación requiere una considerable subjetividad, al no existir una guía clara que determine qué nivel de semejanza (o de diferencia) es suficiente para establecer los diferentes niveles en una clasificación. Durante los ańos 60, muchos científicos fueron atraídos por la fenética numérica, pero hacia mitad de los 70, su popularidad decreció de forma notable por varias razones. En primer lugar y aunque se desarrollaron una gran cantidad de programas para analizar los datos, cuando el mismo conjunto de datos se analizaba con distintos programas producían, con frecuencia, diferentes fenogramas. En segundo lugar los datos se manipulan de tal manera, en la mayoría de los programas, que el ideal de producir un análisis lo menos distorsionado posible resulta inviable. En tercer lugar, los fenogramas producidos tienden a generar grupos polifiléticos al no tener en cuenta la posible “convergencia” de los caracteres, ni considerar las homologías. Por último, muchos “fenetistas” sufrieron el síndrome de la “caja negra”, al desconocer lo que el ordenador estaba haciendo con sus datos. Quizá, uno de los errores más importantes de los “fenetistas” sea el de haber considerado a los organismos como “objetos” inanimados cuando, coincidiendo con G. G. Simpson, “los miembros de un taxón son semejantes porque comparten una herencia común y no se ubican en un taxón porque sean semejantes”. La sistemática cladística (cladismo o sistemática filogenética) tuvo su origen en el libro publicado en 1950 por Willi Hennig (traducción castellana de 1968). Desde entonces su popularidad ha ido en aumento y con los ańos el cladismo ha ido mucho más allá del contexto planteado por Hennig originalmente. La meta del cladismo es producir hipótesis “comprobables” de las relaciones genealógicas entre grupos monofiléticos de organismos. Como metodología está basado completamente sobre la “descendencia común”, o sea, la genealogía estricta. El dendrograma usado por los cladistas es denominado cladograma o árbol (Fig.3) y está construido, únicamente, para mostrar la genealogía, es decir, las relaciones ancestral-descendiente. Como se expresa en el cuadro expuesto anteriormente, el valor principal en los análisis se asigna a las homologías y, de ellas, el peso recae sobre el concepto de caracteres homólogos primitivos frente a derivados o más recientes, identificándose pues las homologías estrictamente como plesiomorfías y apomorfías, respectivamente. Todos los demás conceptos relacionados los definiremos más adelante.
a Apomorfias, Plesiomorfias, Convergencias, paralelismos, evolución regresiva
Un análisis cladístico clásico consta de: la identificación de los caracteres homólogos de los organismos que estemos estudiando; establecimiento de la “dirección del cambio” del carácter, o evolución del carácter (“análisis de la polaridad”); construcción del cladograma de los taxones que poseen el carácter analizado. El paso final debería ser la conversión del cladograma en un esquema de clasificación. Una de las críticas más fuertes que se pueden hacer al cladismo es que el proceso fundamental de la especiación sólo lo considera como la separación de una especie ancestral en dos especies hermanas, a pesar de la existencia de otros numerosos modelos de especiación. Para la cladística, una vez que aparece una especie, se debe iniciar una rama en el cladograma y las dos (o más) líneas representan grupos hermanos, a pesar de que la especie original pueda continuar existiendo. Aceptar esta suposición de trabajo es necesaria para facilitar los métodos de construcción de los cladogramas, como se verá inmediatamente. La sistemática ortodoxa (evolutiva). Tanto la fenética como la cladística han contado, la primera, y cuenta, la segunda, con numerosos seguidores, pero la mayoría de los taxónomos, aún habiendo aceptado los avances de uno o de otro sistema, han conservado la metodología tradicional de la clasificación. Ésta consiste en intentar representar en la clasificación no sólo la ramificación de las líneas filéticas sino también su posterior divergencia y esto se puede hacer indicando en la disposición de distintos taxones si se han vuelto, o no, radicalmente diferentes de los que deberían ser sus grupos hermanos, por la invasión de un nuevo nicho o zona adaptativa. El resultado es la conversión del cladograma en un filograma en terminología de Mayr (1969) o “árbol evolutivo” (fig. 4). El uso del término “evolutiva” es quizá desafortunado pues parece que el utilizar la teoría evolutiva fuese de uso particular para esta filosofía de clasificación, aunque se hace atendiendo a que se siguen los postulados de Darwin en mayor o menor medida. La principal diferencia entre este método y la cladística, está en el considerable peso que se le da a las “autapomorfías” (caracteres derivados adquiridos por un grupo hermano pero no por otro). Frente al término “clade” de la cladística, la sistemática ortodoxa emplea el término “grado” que representa niveles de cambio evolutivo que deben quedar reflejados en la clasificación. Muchos de los contenidos filosóficos de esta metodología sistemática se han expuesto al principio, al comentar el bagaje histórico general de la problemática de la clasificación en Biología. A pesar de lo indicado conviene seńalar que la premisa básica de este método es la teoría de la evolución por selección natural y aunque se ha desarrollado gradualmente, más allá de la tradición linneana, las metodologías o reglas específicas que se utilizan, de forma general, para crear clasificaciones, no han sido nunca expresamente formuladas. Muchos de los procesos se basan en la intuición o en hipótesis, expresadas muy a menudo de forma ambigua, sobre el proceso evolutivo. Por todas estas razones, los “árboles” producidos por este método no se prestan bien para ser comprobados.
No deberíamos finalizar esta primera parte sin volver a referirnos a los aspectos más “cuantitativos” de la Cladística. Los procedimientos metodológicos “numéricos” enfocados bajo una perspectiva filogenética tuvieron como marco fundamental el trabajo de Kluge y Farris (1969), primero de los estudios que trataron de formalizar el uso del “análisis de parsimonia” en la cladística, basada, precisamente, en los conceptos propuestos por Wagner (1961), que era uno de los fenetistas más seńalados. Este estudio generó algunos de los conceptos básicos de los que comenzó a denominarse como “análisis cladístico”, conceptos tales como parsimonia, árboles óptimos, índices de consistencia (CI), y otros, etc. todos ellos se sumaron al arsenal conceptual y metodológico ya propuesto por Hennig. Posteriormente Farris (1970) influyó profundamente en la discusión sobre el método al generalizar el uso de la parsimonia en los análisis no enraizados (sin referencia a la polaridad de los caracteres) y este trabajo acabó de sentar las bases de la cladística numérica. El paso siguiente en este camino fue el desarrollo de varios programas de ordenador que implementaban los análisis de parsimonia, pero, para la sistemática, esto no fue una novedad ya que los programas de análisis sistemático eran comunes entre los fenetistas. La abundancia y optimización de los programas de análisis cladísticos puede hacer parecer que este tipo de análisis, hoy día, es una actividad simple, casi automática. Nada más lejos de la realidad, como seńalan recientemente Ferrarezzi y Marques (1997) “el uso de los métodos numéricos... debe ser mirado exclusivamente, más como una herramienta para ejecutar esa parte del análisis, ahorrando tiempo y proporcionando fiabilidad en cuanto a la precisión de los resultados obtenidos”. Un análisis de las relaciones filogenéticas de un grupo determinado se realiza en dos etapas:
1. Construcción de la lista de caracteres a analizar (que incluye todos los razonamientos anteriores al análisis numérico de la matriz de datos, como son el establecimiento de homologías, su codificación y ordenación, etc.). 2. Elección del árbol óptimo para este conjunto de datos, según el criterio adoptado. Los aspectos críticos de esta “cladística numérica” se pueden sintetizar, siguiendo a los autores antes citados, en las siguientes preguntas:
En el siguiente apartado se tratará de contestar a estas y otras preguntas, así como establecer la metodología básica y los conceptos acerca del tratamiento de los caracteres y de su análisis.
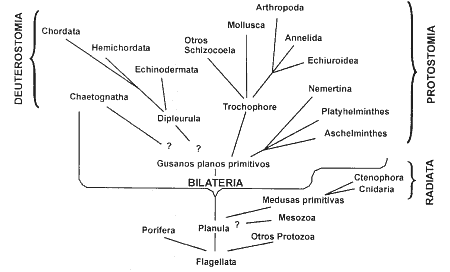
3.- Métodos de reconstrucciones filogenéticas La reconstrucción filogenética se sustenta en el concepto darviniano de "descendencia con modificación", de forma que los caracteres que se observan en las distintas especies, heredados a partir de un ancestro común, son indicativos de una relación genealógica. El postulado del que se parte es que la semejanza puede ser interpretada de forma racional bajo la óptica de una ascendencia común. El problema, por tanto, que plantea la reconstrucción filogenética es el de la inferencia del ancestro a partir de la observación de los caracteres que presentan los taxones terminales. Hasta la aparición de la obra de Hennig (1950), las construcciones filogenéticas se establecían a partir de llamado "triple paralelismo" (Threefold parallelism), consistente en el apoyo de la anatomía comparada, la ontogenia y la paleontología. Su objetivo era encontrar las similitudes o semejanzas entre los taxones basándose en el análisis de los caracteres estudiados bajo este criterio de triple paralelismo, de forma que entre aquellos taxones que más se asemejaran, más próximo era su parentesco. La dificultad estriba en que la semejanza no nos es dada, sino que es interpretada. De los distintos criterios de esta interpretación surgen los diferentes métodos de análisis filogenéticos, y la consecuente controversia entre los biólogos. Desde el punto de vista evolutivo, la semejanza puede ser dividida, simplemente, en homología y en homoplasia. La homología es el parecido heredado a partir de un ancestro común, mientras que la homoplasia es un parecido que no es consecuencia de una herencia común (Simpson, 1961: pág. 78). La homoplasia a su vez se subdivide en convergencia y en reversión. La convergencia, también llamada paralelismo, es la aparición independiente en dos o más especies de un mismo carácter; este carácter no había aparecido antes. La reversión es la aparición, de nuevo, del carácter ancestral, lógicamente este carácter ya había aparecido. Tal como se ha comentado anteriormente, para los sistemáticos taximétricos (Sokal y Sneath, 1963) las relaciones entre taxones solamente pueden ser expresadas basándose en la similitud global manifestada a partir del cálculo de matrices de distancias e índices de similitud, para ello no se hace ningún tipo de distinción entre los caracteres, mezclando caracteres homólogos y homoplásicos. Para los sistemáticos evolutivos (Simpson, 1961; Mayr, 1969) la similitud global propuesta por los taximétricos no puede generar reconstrucciones filogenéticas debido a las falsas similitudes aportadas por las homoplasias. Sólo las similitudes basadas en homologías pueden permitir establecer las filogenias. Para los sistemáticos filogenéticos o cladistas (Hennig, 1950, 1966, 1968; Eldredge y Cracraf, 1980; Wiley, 1981; Nelson y Platnick, 1981) el concepto de homología debe diferenciarse en plesiomorfía (estado primitivo) y apomorfía (estado derivado), de forma que sólo el compartir apomorfías (sinapomorfías) por distintas especies, es indicativo de un parentesco común y los grupos así constituidos son monofiléticos. En la actualidad dos son los campos de actuación en las reconstrucciones filogenéticas, resultado de sus dos distintas concepciones de la similitud. Una de ellas basada en la similitud global: concepción fenética, y la otra basada en el análisis de los caracteres, diferenciados en plesiomorfos, apomorfos y homoplásicos: concepción cladista.
4.- Definición y criterios de homologías La definición de homología es clara y concisa, por homología se entiende todo aquello que se hereda a partir de un ancestro común, de forma que un carácter compartido por diferentes especies es homólogo si se heredó a partir de un ancestro común, compartido por dichas especies. En todo momento debe recordase que la homología es una hipótesis sobre la genealogía. Así, uno puede reconocer una semejanza en determinados caracteres morfológicos mediante la anatomía comparada, y establecer la deducción evolucionista de que tal observación es debida a una ascendencia común. Por tanto, la homología no se observa, sino que se propone una hipótesis de homología a partir de una observación. Tal como expresa Fitch, en Lewin 1987: " Lo importante es distinguir entre la observación y la conclusión". La definición de homología y el criterio de reconocimiento de homología son esencialmente diferentes. Si bien la definición de homología es del todo explícita, se plantea el problema de żCómo reconocer la homología sin conocer a priori la filogenia?. El criterio para establecer la homología es triple (Patterson, 1982): criterio de semejanza, de no-coexistencia y de congruencia. El criterio de semejanza, íntimamente ligado al principio de las conexiones o de idéntica posición, utilizado en la anatomía comparada: un órgano es homólogo en dos o más especies si con independencia de su forma o función posee idénticas conexiones con otros órganos. El criterio de la no-coexistencia, permite distinguir la verdadera homología de la llamada homología seriada: dos caracteres genealógicamente homólogos no pueden existir en un mismo organismo. Patterson lo expone en un ejemplo didáctico sobre los ángeles: “la teoría según la cual el brazo del hombre y las alas de las aves son genealógicamente homólogos, debería ser rechazada si los ángeles fuesen entidades reales, pues presentan a la vez brazos y alas”. El criterio de congruencia que permite superponer los mismos árboles construidos a partir de diferentes caracteres. Los caracteres homólogos son congruentes. Este principio de congruencia se sustenta en el principio de parsimonia.
5.- El método cladista 5.1. El principio de parsimonia El método cladista puede ser calificado como un método hipotético deductivo. La hipótesis versa sobre el sentido de las transformaciones de los caracteres, y las deducciones sobre las afinidades filogenéticas caracterizadas por el método. El principio de parsimonia (Edwards y Cavalli-Sforza, 1963, 1964) es el criterio metodológico adoptado por el cladismo para decidir, en un análisis filogenético que presenta incongruencias entre los caracteres, cual de los cladogramas posibles, el llamado árbol más corto, constituye la reconstrucción más probable que es representativa de la filogenia del grupo. El principio de parsimonia sostiene que la explicación más simple es preferible a las más complejas, es decir, aquella que requiera un menor número de cambios evolutivos, permitiendo tanto las reversiones como las convergencias de los caracteres. Este principio no sólo es de aplicación en análisis cladistas (Camin y Sokal, 1965; Kluge y Farris, 1969) sino también en análisis de distancias (Cavalli-Sforza y Edwards, 1967; Fitch y Margoliash, 1967). La asunción del criterio de la parsimonia de que las transiciones en un carácter son intrínsecamente poco probables, es consecuencia de la tacańería del cambio evolutivo.
Este principio cumple con la finalidad a la que todo análisis filogenético debe estar sometido: la construcción de un esquema racional, y por tanto, no arbitrario. Tal esquema debe estar en todo momento disponible a su refutación o a su confirmación, al introducir nuevos caracteres o taxones. El árbol más corto es el que nos va a permitir tal tipo de control (Darlu y Tassy, 1993: pp 44).
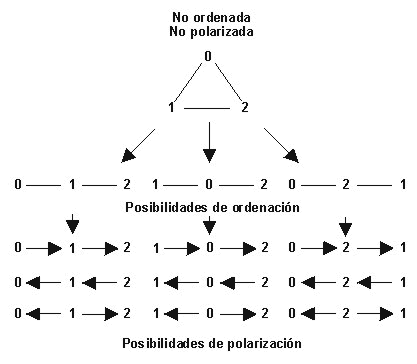
5.2. Codificación, ordenación y polarización de los caracteres La codificación es simplemente la aplicación de un símbolo, generalmente numérico, para cada estado diferente de un determinado carácter o serie de transformación. Los caracteres se codifican de forma que puedan ser analizados comparativamente. En el caso de caracteres con más de dos estados, su codificación puede además contener informaciones adicionales sobre sus conexiones evolutivas; en este caso diremos que los caracteres están ordenados. Si a esta ordenación se le asigna una orientación del cambio evolutivo, la serie de transformación estará polarizada, estableciéndose la relación apomórfica y plesiomórfica relativa de los estados de los caracteres. Al contrario de lo que frecuentemente se piensa, la incorporación de esta información de direccionalidad evolutiva en las series de transformación mediante la polarización a priori de los caracteres es, en general, totalmente innecesaria en el análisis cladista, salvo en casos de imposición de determinadas restricciones del criterio de parsimonia.
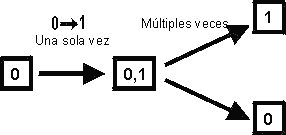
5.2.1.- Caracteres binarios Aquellos que están constituidos por dos estados. Son los de
codificación más simple pues automáticamente constituyen un par ordenado,
representado bajo la forma de 0-1 o a-b, en el que uno de ellos representa el
estado plesiomorfo y el otro el apomorfo, pero a priori no indican una
orientación particular. Las transformaciones a
5.2.2.- Caracteres multiestado Los caracteres multiestado son aquellos que presentan más de dos estados en su serie de transformación, y cuyas relaciones o conexiones de transformación de un estado a otro pueden ser de dos tipos:
5.3. Modelos de parsimonia Aunque los métodos de parsimonia siempre buscan minimizar el número de pasos en las transformaciones de un carácter, existen distintas modalidades de parsimonia que difieren con relación a las restricciones que se imponen a dichas transformaciones, afectando a la topología final del árbol así como a su longitud total.
5.4. Búsqueda del árbol más parsimonioso En los apartados precedentes se ha visto que existen distintos tipos de métodos que pueden ser elegidos para realizar un análisis numérico de los datos, tales como ordenar o no ordenar los estados de los caracteres, escoger un determinado criterio de parsimonia, o también la posibilidad de asignarles un determinado peso. Todos estos criterios influyen en la búsqueda del árbol más parsimonioso (AMP), "most parsimoniosus tree", es decir, aquel o aquellos árboles que minimizan el número de pasos requeridos para explicar una matriz o conjunto de datos. El problema es que con n taxones terminales se pueden construir (2n-3)! /[2n-2 (n-2)!] árboles dicotómicos (para n=10 existen aproximadamente 3,5 x 107 árboles). De ello se concluye que la comparación de tal número de árboles resulta una operación tediosa, sino imposible.
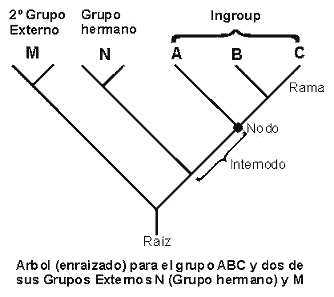
5.5. Polarización por grupo externo. Un grupo externo es un taxón, o grupo de taxones, filogenéticamente emparentado con el grupo en estudio. La polarización por el método del grupo externo pretende la reconstrucción de un ancestro hipotético, ya que la identificación previa de un "verdadero" ancestro en todo análisis filogenético raramente se encuentra. La polarización por comparación con el grupo externo es simple y clara: dado un grupo monofilético, se infiere que el estado ancestral más parsimonioso es aquel que acontece en el grupo externo, pues cumple la hipótesis de parsimonia de exigir un menor número de pasos para explicar la transformación de los estados de los caracteres en apomorfos y plesiomorfos, entre los componentes del grupo interno. El problema práctico es que entre los caracteres del grupo(s) externo(s) elegido y el grupo interno surgen incoherencias o contradicciones filogenéticas. Para minimizar este problema Maddison et al. (1984) propone analizar los grupos externos de forma separada al del grupo interno, tratando de optimizar los caracteres y asumiendo una relación previa entre los grupos externos. Las etapas de esta aplicación consisten en asumir el monofiletismo del grupo de estudio o interno, definiendo dos nodos: uno correspondiente al grupo interno y que representa el ancestro común más reciente al grupo interno y otro, el llamado nodo del grupo externo, que corresponde al ancestro común relativamente más antiguo común al grupo interno y a uno de los grupos externos más próximo. A partir del establecimiento de estos nodos y del conocimiento de las relaciones existentes entre los grupos externos entre sí y con el grupo interno, se define la topología de los grupos externos. El problema de este método es que si no se resuelven o conocen las relaciones entre los grupos externos y de éstos con el grupo interno, puede ser imposible inferir la polaridad de los caracteres. Por tanto, la polarización de los caracteres como paso inicial en el análisis cladista, además de ser innecesario, puede conducir a resultados que no sean globalmente parsimoniosos, por pretender validar una parsimonia en dos fases sucesivas y disyuntas (Nixon y Carpenter, 1993; Ferrarezzi y Marques, 1997).
5.6. Enraizamientos y grupo(s) externo(s). Una forma más segura y práctica de evitar los problemas anteriormente mencionados, es la inclusión de todos los taxones en el análisis filogenético, dejando que los ancestrales hipotéticos sean dilucidados a posteriori como resultado de la aplicación de dicho análisis. Esto se debe a que la longitud de un árbol es independiente de la posición de la raíz o de la elección de cualquier brazo terminal como grupo externo, no siendo necesaria la polarización previa de los caracteres. El enraizamiento y la consecuente polarización de los caracteres se obtendrá en un punto intermedio entre el, o los, grupo(s) externo(s) y el grupo interno. Lundberg (1972) propone otro método de enraizamiento, resolviendo en primer lugar la topología del grupo interno a través de un análisis de Parsimonia no enraizada y posteriormente valida cual es la posición en la que un ancestro hipotético puede ser insertado de forma más parsimoniosa al árbol obtenido. Se aconseja la aplicación de este método en aquellos casos en que no es posible obtener grupos externos adecuados (Nixon y Carpenter, 1993).
5.7. Medidas de la homoplasia Al comparar distintos árboles, éstos pueden diferir en su diseńo. La decisión para establecer cual de esos árboles es el más representativo, el por qué lo es y en que medida, puede ser argumentada a partir de la medida de la cantidad de información filogenética contenida en los árboles y obtenida mediante el estudio de una serie de índices que muestren la carga de homoplasia.
HER = (M-L) / (M-R)
HERM = (G-L) / (G-R)
5.8. Árbol de consenso El árbol de consenso trata de resolver el problema de la comparación entre árboles que tienen una misma longitud o igualmente parsimoniosos, tratando de optimizar las diferentes topologías y no de optimizar la matriz de datos. La búsqueda de este árbol, se condensa en una única representación en la que se diferencian claramente las partes concordantes entre los distintos árboles, de las de las partes discordantes. Los métodos de consenso más generalizados son: el método estricto o de Nelson (Sokal y Rohlf, 1962, 1981; Nelson, 1979), semiestricto (Bremer, 1990), de Adams (Adams, 1972) y de consenso mayoritario (Margush y McMorris, 1981). Estos métodos difieren en el criterio por el que se forman los subgrupos de taxones en el árbol de consenso. En general, el árbol de consenso de un conjunto de árboles es menos resolutivo, de mayor longitud, que los árboles iniciales y menos informativo. Ello se debe al hecho de que los caracteres sobre el árbol de consenso tienden a cambiar más veces que en un árbol cualquiera, en que los cambios se minimizan. Debe destacarse que ningún árbol de consenso es la hipótesis más idónea para el conjunto de los datos, sino que debe interpretarse como el grado de acuerdo o desacuerdo entre varios árboles obtenidos después de una búsqueda y no como un árbol filogenético.
6. El método de compatibilidad El método de compatibilidad es, en cierta medida, una variante de los métodos de parsimonia, pues si bien utiliza el principio de parsimonia, se diferencia de ellos en cómo trata la homoplasia. La compatibilidad se basa en los trabajos de Le Quesne (1969, 1972), en los que define los caracteres mutuamente compatibles como aquellos cuyos cambios de estado sobre un árbol determinado pueden ser explicados sin recurrir a la homoplasia, es decir, que el árbol sólo presenta una transformación por carácter. Al conjunto de caracteres mutuamente compatibles se les denomina "clique" (Estabrook et al., 1977). Los caracteres que no son compatibles son, por tanto, homoplásicos. El método de compatibilidad consiste simplemente en buscar el árbol para el cual el "clique" es mayor, despreciando los caracteres que son homoplásicos. El rechazo de este método a usar caracteres homoplásicos en la búsqueda del árbol filogenético connlleva una serie de problemas. Así, puede ser que existan caracteres sometidos a homoplasia, pero que sin embargo sea diagnóstico de un grupo monofilético situado en el interior del grupo estudiado. Su supresión supondrá la pérdida de una información filogenéticamente útil. Por ejemplo, la homeotermia se considera una sinapomorfía, por una parte en las aves y, por otra, en los mamíferos. Este carácter ha aparecido dos veces por convergencia. Si esta ocurrencia es correcta, el carácter homeotermia no será considerado en un análisis de compatibilidad entre los amniotas, rehusando explicar como ha aparecido este carácter y suprimiendo información filogenética útil.
Bibliografía Adams, E. N., 1972. Consensus techniques and the comparison of taxonomic trees. Syst. Zool., 37: 27-29. Archie, J. M., 1989 a. A randomization test for phylogenetic information in systematic data. Syst.Zool., 38(3): 239-252. Archie, J. M., 1989 b. Homoplasy excess ratios: new indices for measuring levels of homoplasy in phylogenetic systematic and a critique of the consistency index. Syst. Zool., 38(3): 243-269. Bremer, K., 1990. Combinable component consensus. Cladistics, 6: 369-372. Brusca, R.C. & Brusca, G. J., 1990. Invertebrates. Sinauer Associates, Inc. Sunderland, Massachusetts, USA. 922 pp. Camin, J. H. & Sokal, R. R., 1965. A method for deducing branching sequences in phylogeny. Evolution, 19: 311-326. Cavalli-Sforza, L.L. & Edwards, A. W., 1967. Phylogenetics analysis: models and estimation procedures. Am. J. Hum. Genet., 19: 233-257. Chace, F. C., 1969. Unknown species in the sea. Science, 163: 1271 Darlu, P. & Tassy, P., 1993. La reconstrution phylogénétique. Concepts et méthodes. Masson (Paris). 245 pp. Edwards, A.W. & Cavalli-Sforza, L. L., 1963. The reconstrution of evolution. Ann. Hum. Genet., 27: 104-105. Edwards, A.W. & Cavalli-Sforza, L. L., 1964. Reconstrution of evolutionary tree. Systematics Association Publication. 6: 67-76. Eldredge, N & Cracarft, J., 1980. Phylogenetic patterns and the evolutionary process. Columbia University Press. New York. Estabrook, G. F., Strauch, J. G. & Fiala, J. K., 1977. An aplication of compatibility analysis to the Blackiths' data on orthopteroid insects. Syst. Zool., 26: 269-276. Farris, J. S., 1970. Methods for computing Wagner trees. Syst. Zool., 19: 83-92 Farris, J. S., 1977. Phylogenetics analysis under Dollo's law. Syst. Zool., 26: 78-88. Farris, J. S., 1978. Inferring phylogenetics trees from chromosome inversion data. Syst. Zool., 27: 275-284. Farris, J. S., 1989. The retention index and homoplasy excess. Syst. Zool., 38(4): 406-407. Felsenstein, J., 1979. Alternative methods of phylogenetic inference and their interrelationship. Syst. Zool., 28: 49-62. Ferrarezzi, H. & Marques, A.C., 1997. Anŕlise cladística numérica e recursos computacionais. 164-186. En: Amorin, D. de S. (Ed.) Elementos Básicos de Sistemática Filogenética (2Ş ed.). Holos Editora y Sociedade Brasileira Entomología, Riberăo Preto y Săo Paulo. Brasil. 276 pp. Fitch, W. M., 1971. Distinguishing homólogous from analogous proteins. Syst. Zool., 20: 406-416. Fitch, W.M. & Margoliash, E., 1967. Constrution of phylogenetic trees. Science, 155: 279-284. Hendy, M.D. & Penny, D., 1982. Branch and bound algorithms to determine minimal evolutionary trees. Math. Biosci., 59: 277-290. Hennig, W., 1950. Grundzüge einer Theorie der Phylogenetischen Systematik. Deutscher Zentralverlab, Berlin. 370 pp. Hennig, W., 1966. Phylogenetic Systematics. University of Illinois Press. 263 pp. Hennig, W., 1968. Elementos de una sistemática filogenética. Editorial Universitaria de Buenos Aires. Buenos Aires, Argentina. 353 pp. Huxley, J., (ed.), 1940. The New Systematics. Clarendon Press., Oxford, 583 pp. Huxley, J., 1942. Evolution, the modern synthesis. Allen and Unwin, London, 645 pp. Kluge, A.G. & Farris, J. S., 1969. Quantitative phyletics and evolution of anurans. Syst. Zool., 18(1): 1-32. Le Quesne, W.J., 1969. A method of selection of characteres in numerical taxonomy. Syst. Zool., 18(2). 201-205. Le Quesne, W.J., 1972. Further studies based on uniquely derived character concept. Syst. Zool., 21(3): 281-288. Lewin, R., 1987. When does homology mean something else? Science, 237:1570. Lundberg , J. G., 1972. Wagner networks and ancestors. Syst. Zool., 21: 398-413. Maddison, W. P., Donoghue, M. H. & Maddison, D. R., 1984. Outgroup analysis and parsimony. Syst Zool., 33:83-103. Margush, T. & McMorris, F. R., 1981. Consensus n-trees. Bull Math. Biol., 43: 239-244. Mayr, E., 1969. Principles of Systematic Zoology. McGraw-Hill, New York, 428 pp. Mayr, E., 1982. The Growth of Biological Thought. Harvard Univ. Press., Cambridge, Mass. 974 pp. Mayr, E., Linsley, E.G. & Usinger, R.L., 1953. Methods and Principles of Systematics Zoology. McGraw-Hill, New York, 328 pp. Nelson, G., 1979. Cladistic analysis and synthesis: Principles and definitions, with a historical note on Adamson´s Familles des Plantes (1763-1764). Syst. Zool., 28: 1-29. Nelson, G. & Platnick, N., 1981. Systematic and biogeography: cladistics and vicariance. Columbia University Press, New York. 567 pp. Nixon, K.C. & Carpenter, J. M., 1993. On outgruops. Cladistics, 9: 413-426. Patterson, C., 1982. Morphological characters and homology. 21-74. In: Joysey, K.A. & A.F. Friday (Eds). Problems of phylogenetic reconstruction. Academic Press. London. 442 pp. Savory, T., 1970. Animal Taxonomy. Heinemann Educational, London, 101 pp. Simpson, G. G., 1961. Principles of Animal Taxonomy. Columbia Univ. Press., New York, 247 pp. Sneath, P. H. A. & Sokal, R. R., 1973. Numerical Taxonomy. The principles and practice of numerical classification. Freeman, San Francisco, California. 573 pp. Sokal, R. R. & Rohlf, F. J., 1962. The comparison of dendograms by objective methods. Taxon, 11: 33-40. Sokal, R. R. & Rohlf, F. J., 1981. Taxonomic congruence in the Lepopodomorpha reexamined. Syst. Zool., 30: 309-325. Sokal, R.R. & Sneath, P. H. A.,1963. Principles of numerical taxonomy. W. H. Freeman and Co. San Francisco. 359 pp. Strickland, H. E., 1840. Observations upon the affinities and analogies of organized beings. Ann.Mag.Nat. Hist., 4: 219-226. Swofford, D. L. & Olse, G. J.,1990. Phylogeny reconstruction. In: Hillis D. M. & Moritz, C. (Eds). Molecular Systematic. Sinauer Ass. (Sunderland, Massachusetts): 411-501. Wagner, W. J. Jr., 1961. Problems in the classification of ferns. In: Recent advances in Botany. University of Toronto Press., Montreal: 841-844. Wiley, E. O., 1981. Phylogenetics. Theory and practice of phylogenetic systematic. John Wiley and sons. New York. 439 pp.
|
|
© 1999-2001 SEA, Sociedad Entomológica Aragonesa http://entomologia.rediris.es/sea - CV-e Comunidad Virtual de Entomología - http://entomologia.rediris.es - admin@entomologia.rediris.es |